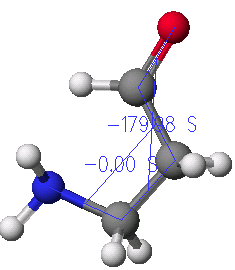
그러면 좌측에 도구막대(toolbar)가 나오게 되는데 selection tool, drawing tool, manipulation tool로 3가지로 구성되어 있고 상단에는 메뉴(Menu)와 기능관련 도구막대 그리고 리본(ribbon)이 있습니다.
선택도구(selection tool)
![]() 원자, 결합을 선택할 때 사용하는 버튼입니다.
원자, 결합을 선택할 때 사용하는 버튼입니다.
![]() 분자를 선택할 때 사용하는 버튼입니다.
분자를 선택할 때 사용하는 버튼입니다.
![]() 미리 선택된 부분과 유사한 부분(fragment)를 모두 선택할 때 사용하는 버튼입니다.
미리 선택된 부분과 유사한 부분(fragment)를 모두 선택할 때 사용하는 버튼입니다.
그리기도구(drawing tool)
![]() 분자를 그릴 때 사용하는 버튼입니다.
분자를 그릴 때 사용하는 버튼입니다.
조정도구(manipulation tool)
![]() 평면상, 공간상 에서 회전을 시킬 때 사용하는 버튼입니다.
평면상, 공간상 에서 회전을 시킬 때 사용하는 버튼입니다.
![]() 평면상에서 이동할 때 사용하는 버튼입니다.
평면상에서 이동할 때 사용하는 버튼입니다.
![]() 확대 또는 축소를 할 때 사용하는 버튼입니다.
확대 또는 축소를 할 때 사용하는 버튼입니다.
기능관련 도구막대
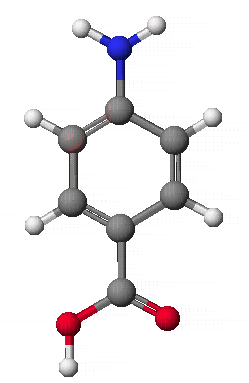
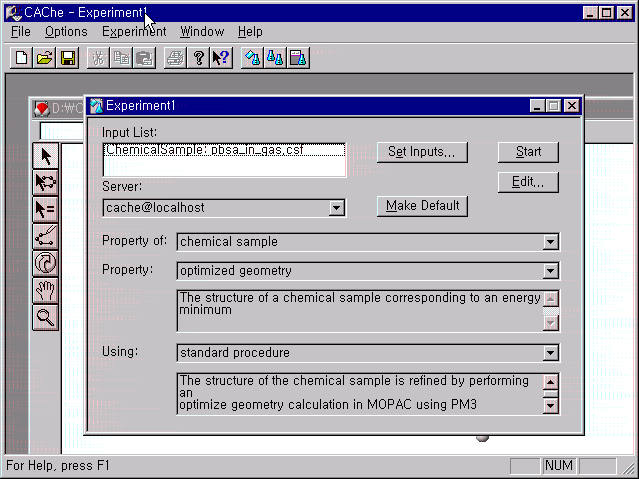
먼저 예로 ρ-aminobenzoic acid(PABA)의 화학구조를 그려보도록 합시다.
ρ-aminobenzoic acid(PABA)는 적혈구 형성에 도움을 주며, 머리카락 색을 자연색을 바꾸어 주는 역할을 하고, 공업적으로는 태양선 차단제로 사용되고 있습니다. ρ-aminobenzoic acid(PABA)가 생체내에서 부족할 경우 극도의 피곤과 습진, 두통과 우울증등을 유발하며 조기에 머리카락색이 희게 되는 현상을 일으키기도 합니다.
먼저 그리기 도구를 이용하여 분자를 그릴 때 원자의 종류와 결합종류 그리고 전하량과 혼성화(hybridization)등을 확인하여야 합니다. 일반적으로 구조의 골격을 모두 탄소로 그린후 선택도구를 이용하여 원자와 결합종류, 전하량, 혼성화 등을 바꾸어 주는 것이 사용하기에 용이합니다.
그리기 도구인![]() 를 선택한 후 메뉴아래에 나타나는 리본(ribbon)에서 원자의 종류는 C로, 혼성은 sp3 tetrahedron으로, 전하는 0으로, 결합종류는 single로 조정합니다.
를 선택한 후 메뉴아래에 나타나는 리본(ribbon)에서 원자의 종류는 C로, 혼성은 sp3 tetrahedron으로, 전하는 0으로, 결합종류는 single로 조정합니다.
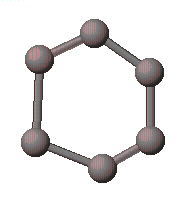

그리기 도구를 선택하여 수소를 제외한 나머지 부분들(amino 와 carboxy group)을 Carbon으로 그립니다.
선택도구인 ![]() 를 선택한 후, 해당하는 원자를 선택한 후에 리본에서 원자의 종류를 각각 Nitrogen과 Oxygen으로 바꿉니다. 이중결합으로 만들어야 하는 부분은 결합의 중간부분을 선택하여 리본에서 결합종류를 Single에서 Double로 바꿉니다.
를 선택한 후, 해당하는 원자를 선택한 후에 리본에서 원자의 종류를 각각 Nitrogen과 Oxygen으로 바꿉니다. 이중결합으로 만들어야 하는 부분은 결합의 중간부분을 선택하여 리본에서 결합종류를 Single에서 Double로 바꿉니다.
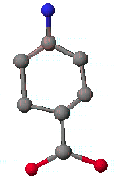
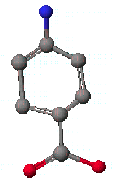
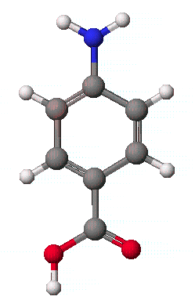
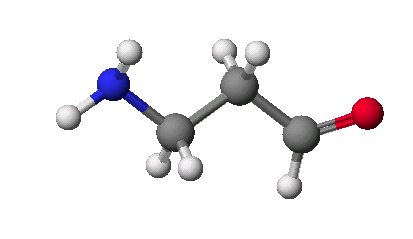
먼저 5개의 탄소를 그린 후 ![]() 를 이용하여 산소와 질소부분으로 바꾸어 주도록 합니다.
를 이용하여 산소와 질소부분으로 바꾸어 주도록 합니다.
기본 골격구조가 그려지면 Beautify 메뉴에서 Comprehensive를 선택하여 위와 같이 적당한 구조형태를 같도록 합니다. 이 과정이 익숙하지 않으시면 1.1.1과정으로 가서 다시 보시기 바랍니다.
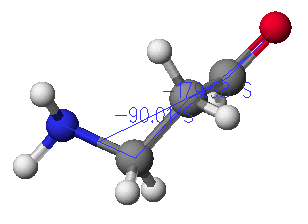
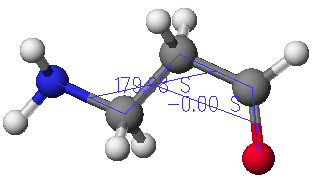
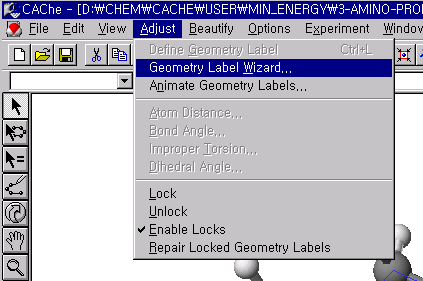
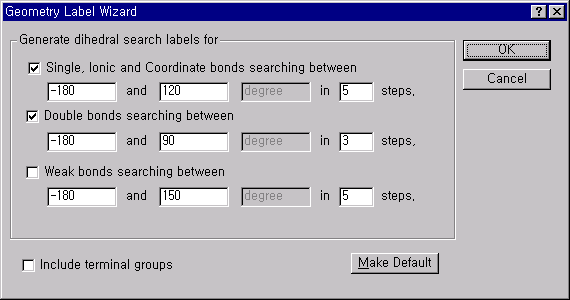
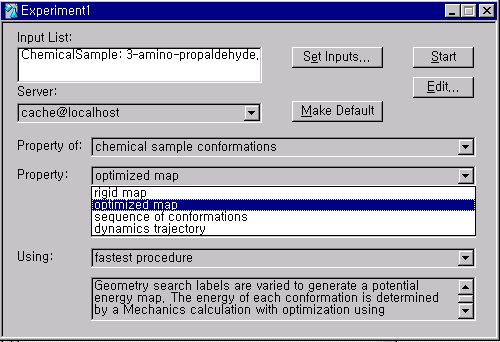
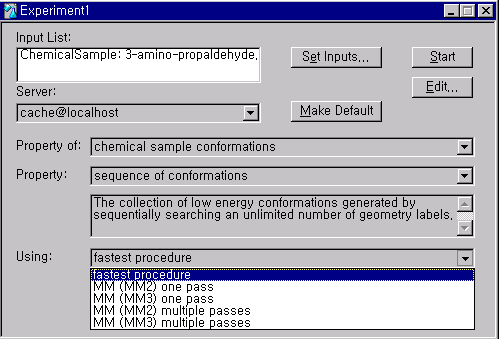
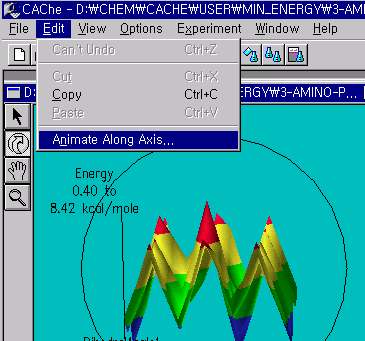
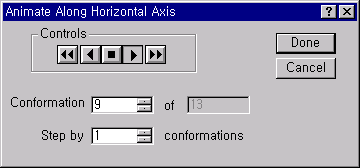
1.2.3. 형태찾기 라벨(Conformational search label) 결정
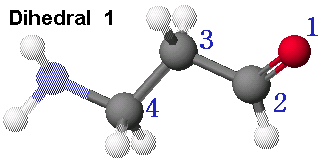
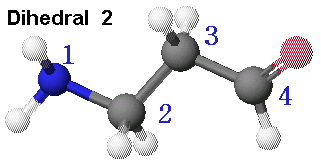
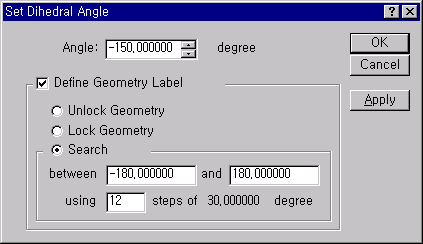
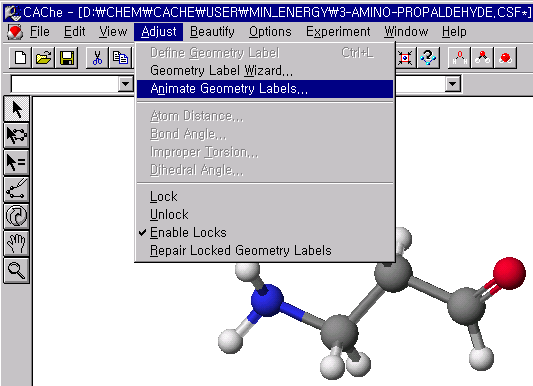
모든 가능한 형태의 구조를 찾기 위해서는 먼저 2개의 탄소-탄소 결합을 회전시킬때의 에너지 변화를 알아볼 필요가 있습니다. 그러려면 회전시킬 2개의 이면각(dihedral angle)를 정의하여야 합니다.