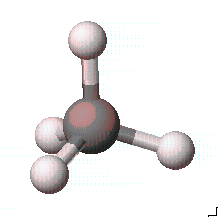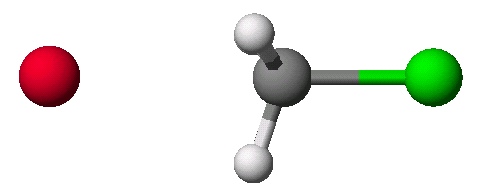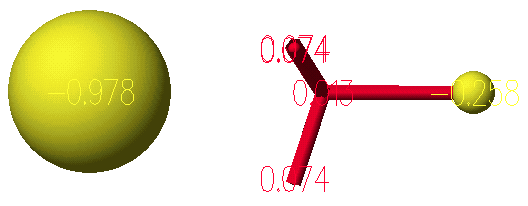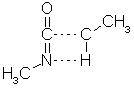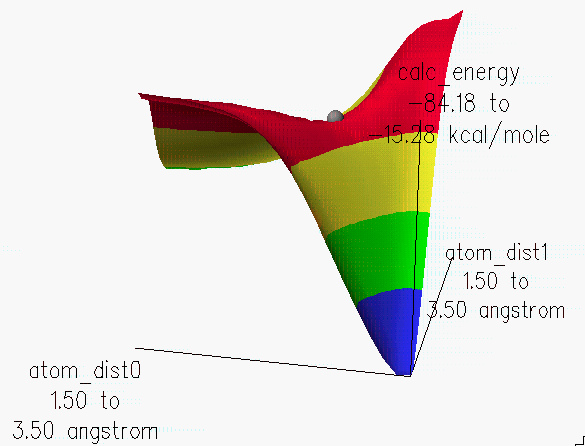본 과정에서는 실제 화합물을 합성하고자할 때의 반응성을 보거나, 반응 경로의 전이 상태 등을 CAChe를 통하여 분자 모델링을 해 보도록 하겠습니다.
3.1 반응성 보기(Reactivity visualization)
본 과정에서는 방향족 화합물에서의 친전자성 치환(Electrophilic substitution)반응에 대하여 치환체 위치에 대한 반응성을 알아보도록 하겠습니다.
3.1.1. 분자 그리기 및 계산
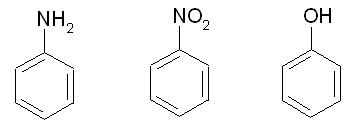
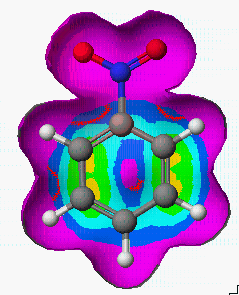
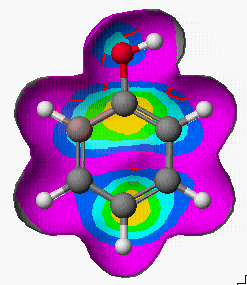
반응성을 보기 위하여 사용한 분자들은 Aniline, Nitrobenzne, Phenol입니다.
먼저 각각의 화합물을 그리고 저장을 합니다. 모든 구조를 항상 새로 그릴 필요 없이 copy & paste를 이용하면 보다 빠르게 작업을 할 수 있습니다.
그 다음 Beautify 메뉴 안의 Comprehensive를 실행시켜 적당한 구조를 만들고, Experiment 메뉴 안의 New를 선택합니다.
Property of는 chemical sample로, Property는 electrophilic susceptibility로, Using은 MM/PM3 geometry with PM3 wavefunction으로 선택합니다. 이 방법은 MM3방법에 이어 PM3방법으로 먼저 분자구조를 최적화 시키고 난후에, PM3의 wavefuction으로 electrophilic attack이 가능한 부분을 3차원 등고선을 통해 시각적으로 보여줍니다.
이 때 Property에는 다양한 분자의 특성을 나타낼 수 있는데, 다음과 같습니다.
UV/Visable transitions
IR transitions
HOMO & LUMO
HOMO-5 & LUMO+4
all molecular orbitals
electron density
electrostatic isopotential
electrostatic potential on electro density
electrophilic susceptibility
nucleophilic susceptibility
radical susceptibility
superdelocalizability-E
superdelocalizability-N
superdelocalizability-R
3.1.2. 부분 전하 보기(Visualization of Partial Charge)
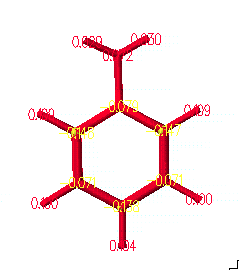
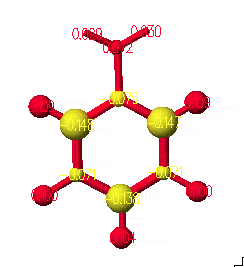
일단 먼저 aniline의 계산이 마치면, 분자가 그려져 있는 원도우로 돌아가서 View 메뉴 안의 Partial Chg & Calc Bond Order를 선택하여 그래픽으로 부분 전하(partial charge)와 결합 차수(bond order)를 볼 수 있습니다. 음전하는 노란색으로 양전하는 빨간색으로 표현되며, 결합차수도 나타납니다.
물론 친전자성 민감도(electrophilic susceptibility)를 계산하였지만, 앞에서 설명했던 것처럼 semi-empirical방법으로 계산을 했을 경우에는 전하와 결합 차수는 자동적으로 결과를 얻게 됩니다.
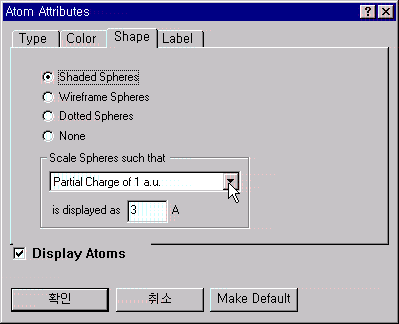
전하의 크기를 그래픽으로 보기를 원할 때는 View 메뉴 안의 Atom Attributes를 선택하고, 아래 왼쪽에 있는 원도우가 나타나면 Shape panel에 가서 Shaded Spheres와 Partial Charge of 1 a.u.로 선택하고 3Å으로 써줍니다. 이것은 전하의 원자단위 1이 3Å크기의 원으로 표현하도록 한 것입니다.
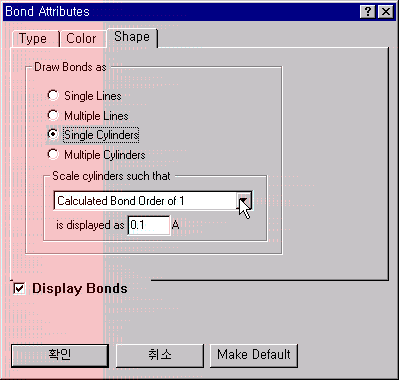
또한 원자간 선의 두께는 결합차수를 나타내게 되는데, View 메뉴 안의 Bond Attributes를 선택하고, 위의 오른쪽과 원도우가 나타나면 Shape panel에 가서 Single Cylinder와 Calculated Bond Order of 1로 선택하고, 0.1Å으로 써줍니다. 이것은 계산된 결과에서 결합 차수가 1일 때의 두께를 0.1Å로 표현하도록 한 것입니다.
위에서의 결과 그림과 같이 벤젠 링(ring)안에 있는 모든 결합은 두께가 같으며, 탄소와 수소와의 결합보다 약 1.5배 더 굵은 것을 알 수 있습니다. 이것은 단지 방향족성(aromaticity)이나 콘쥬게이션(conjugation)을 나타내는 것 뿐아니라 계산된 결합 차수는 전이상태 구조(transition state geometry)의 원자간 결합의 차수를 결정 지을 때도 유용하며, 이것은 뒤에서 설명하도록 하겠습니다. 일단 다시 View 메뉴 안의 Ball and Cylinder를 선택하여 구조를 원래 모습으로 바꾸고 각각 저장하도록 합니다.
3.1.3. 친전자성 민감도 보기(Visualization of Electrophilic Susceptibility)
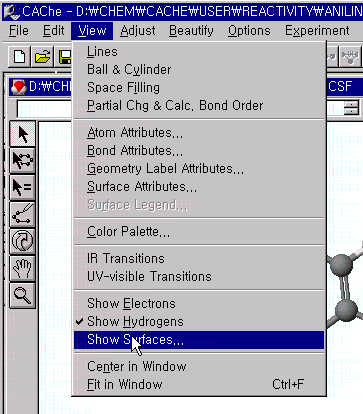
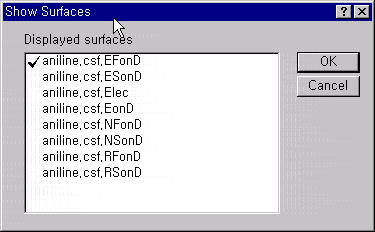
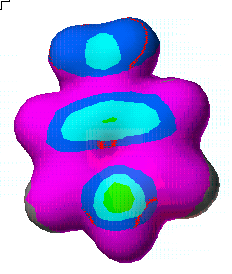
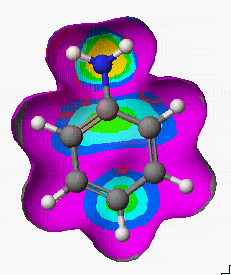
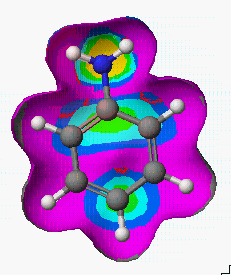
Aniline의 이미 계산된 친전자성 민감도(Electrophilic Susceptibility)를 3차원 등고선으로 보기 위해서는 View 메뉴에서 Show surface를 선택하고, 그림에서 처럼 aniline.csf.EFonD를 선택하면 됩니다.
그 밖에 다른 특성들을 계산 했을 때,
파일명.NFonD– nucleophilic susceptibility
파일명.RFonD– radical susceptibility
파일명.ESonD — superdelocalizability-E
파일명.NSonD– superdelocalizability-N
파일명.RSonD– superdelocalizability-R
파일명.Elec — electrostatic isopotential
파일명.EonD — electrostatic potential on electro density
를 나타내므로 선택하여 속성을 볼 수 있습니다.
이미 계산을 한 상태에서 다른 속성을 계산할 때는 앞에서와 같이 MM/PM3 geometry with PM3 wavefunction로 계산하면 MM에 의해 구조 변화가 생기므로 전단계에서 최종적으로 계산한 PM3 Geometry with PM3 wavefunction으로 계산하여야 합니다.
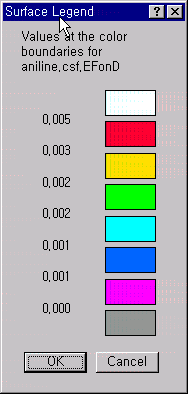
여기서 결정되어 지는 표면적은 0.01 e-/Å3의 전자 확률 민도를 갖는 부분으로 결정되어 지며, 이 표면적 위에 크기에 따라 각각의 색을 표현되게 됩니다.
나타난 등고선의 색에 대하여 값의 크기를 보려면 View 메뉴안의 Surface Legand를 선택하면 색이 나타내는 값을 알 수 있습니다.
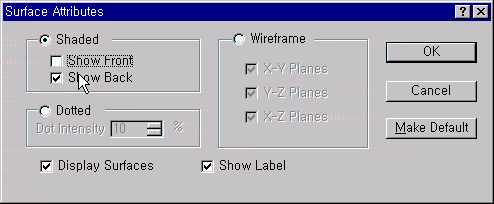
또한 View 메뉴의 Surface Attributes를 선택하여 Shaded를 Show Back으로 나타내면, 분자구조와 표면적 속성을 동시에 볼 수 있습니다.
그렇다면 aniline, nitrobenzene, phenol들의 친전자성 치환(Electrophilic substitution)반응에 있어서 치환체 위치에 대한 반응성은 어떻게 비교 할 수 있을까요?
Aniline Nitrobenzene Phenol
실험 경향 : p, o > m m > o > p p, o > m
실제 반응성에 대한 실험적인 연구결과와 비교해 보면, 대략적으로 일치함을 알 수 있습니다.
또한 등고선의 분포에 따라서 Nitrobenzene > Phenol > Aniline 순으로 친전자성 치환(Electrophilic substitution)반응이 일어날 것으로 예상됩니다.
이렇게 표면적에 나타내는 속성들은 직접적으로 화합물을 합성하기 전에 반응물의 반응성을 점검하고 반응 진행을 예상할 수 있다는 장점을 지니고 있습니다.
3.2 전이상태 모델링(Transition state modeling) – Kinetics & Thermodynamics
전이상태(transition state)를 결정하는 과정은 크게 4가지방법으로의 접근이 있습니다.
순서에 따라 점차로 계산시간이 증가하게 배열하였습니다.
◎ 전이상태로 예상되는 구조를 예상한 후에 그 구조를 정교화(refine)하는 방법
◎ 전이상태를 알고 있는 유사한 반응을 변형하여 그 구조를 정교화(refine)하는 방법
◎ Saddle 계산을 수행하는 방법 (MOPAC)
◎ 최적화된 격자(Optimized grid) 또는 반응좌표(reaction coordinate) 방법 (MOPAC) : 이방법은 어려운 전이상태를 찾는데 있어서 강력한 방법입니다. 그밖에 다른 방법들도 논문이나 책을 참고하시길 바랍니다.
그리고 전이상태를 찾는 계산은 PM3와 AM1, ZINDO와 같은 semi-empirical방법이나 양자화학적 방법으로만 가능합니다.
3.2.1. 예상구조에서 전이상태를 찾기 위한 구조 그리기
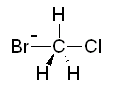
첫 번째로 예상한 전이상태를 refine하는 과정입니다. 실제 전이상태에 가까운 구조를 그릴수 있다면, 쉽게 해결할 수 있는 방법입니다. 먼저 methyl chloride와 Br–와의 SN2반응을 예로 들어 시작하겠습니다.
SN2 반응 : methyl chloride 와 Br–
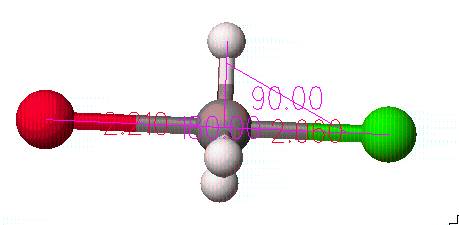
기본적으로 분자를 그리는 과정은 먼저 구조가 위의 그림처럼 탄소가 5개의 원자들과 결합되어 있는 형태로 그려져야 합니다.
탄소 하나를 그린 후에 Beautify Comprehensive를 실행합니다.
4개중 하나의 수소를 염소로 바꾼 뒤에 다시 Beautify Comprehensive를 선택합니다.
그리기 도구를 이용하여 Br를 그리고 난 후에 Br만을 선택하고 위의 그림처럼 전하를 -1로 해주고 Beautify geometry를 선택하여 Br과 C와의 거리만을 알맞게 바꾸어주도록 합니다. Br만 선택하지 않고 Beautify geometry를 실행하거나 Beautify Comprehensive를 실행할 경우, 전이상태와 비슷한 구조로 갈 수 없기 때문에 주의해야 합니다.
실제 전이상태에 해당하는 구조를 가지도록 만들려면, 임의로 구조를 바꾸어 주어야 합니다.
Br-C-Cl는 180도 이고, 수소는 이것과 수직이어야 합니다. 각각 3개의 원자를 선택하고 Adjust 메뉴안의 Bond angle을 선택하여 해당하는 각을 넣어줍니다.
이때 원자를 선택하는 순서는 앞에서 언급한데로 첫 번째로 선택하는 원자가 이동하므로 구조를 고려하여 결정하여야 합니다.
그다음 Cl과 C와의 거리, 그리고 Br과 C와의 거리를 늘려야 하는데, 현재 상태보다 각각 0.3Å씩 바깥쪽으로 이동하도록 합니다. 이 때도 먼저 선택한 원자가 움직이므로 Cl이나 Br을 먼저 선택해야합니다. 최종적으로 만들어진 구조는 아래와 같습니다.
3.2.2. 예상구조에서 전이상태 계산
계산을 하기 앞서서 저장을 하여야 합니다. 이때 그려진 화학구조의 hybridization과 valence가 맞지 않는 원자를 가지고 있는 주의가 나타나게 되는데 이것은 Br의 전하가 -1이기 때문에 valence가 맞지 않는다고 나오는 메시지입니다. 이때는 아니오를 선택하고 sn2라는 이름으로 저장합니다.
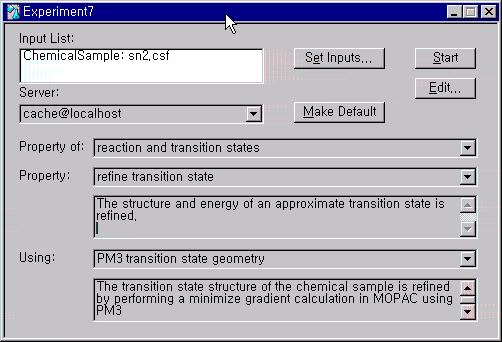
Experiment안에 New를 선택하고, 다음 페이지의 원도우가 나타나면, Property of는 reaction and transition states로, Property는 refine transition state로, Using은 PM3 transition state geometry로 선택하고 계산을 시작합니다
일반적으로 Optimized Geometry는 초기구조에서 먼저 퍼텐셜 에너지를 계산하고, 에너지가 낮아지는 방향으로 원자 위치를 변화시키는 과정입니다. 원자 위치의 변화와 방향은 퍼텐셜 에너지 표면의 기울기에 의존하여 진행되고, 이러한 과정은 변화된 구조의 에너지 차이(에너지의 1차 미분값)가 일정한 한계만큼 작아졌을 때까지 반복 수행됩니다. 그러나 전이상태를 결정하는 refine transition state의 경우 구조변화를 에너지 차이가 작아지는 지는 지점으로만 이동하게 되어 있으므로 초기구조가 전이 상태에 가까운 구조이어야 합니다.
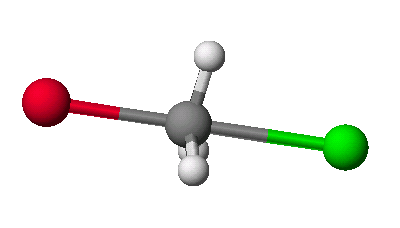
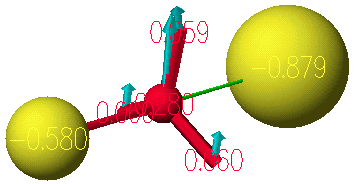
계산이 끝나면 위와 같은 구조를 나타낼 것이며, 전이상태의 생성열(heat of formation)이 대략 -65kcal/mol이 나오는 것을 확인합니다. 그리고 타당한 전이상태인지를 확인하기 위하여 그 구조의 부분 결합 차수(partial bond order)를 보도록 합니다.
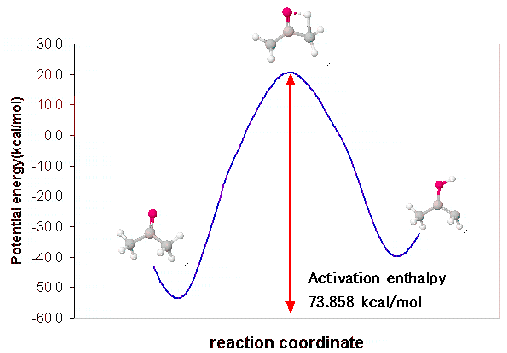
반응물에서 전이상태까지의 활성화 엔탈피(activation enthalpy)를 결정하기 위해서는 반응물의 생성열(heat of formation)을 계산할 필요가 있습니다. 이렇게 하기 위해서는 하나의 원도우에 두 반응물을 그리고 PM3방법으로 Optimized geometry를 실행합니다.
활성화 엔탈피(activation enthalpy)는 -64.80672 – (-81.10152) = 16.2948 Kcal/mol로 계산됩니다.
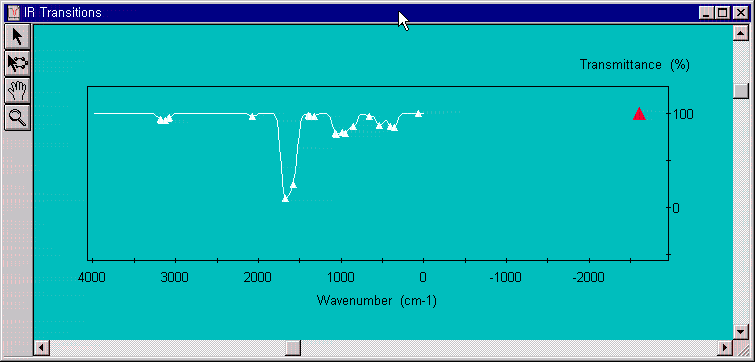
3.2.3. IR 스펙트럼으로 전이상태 확인
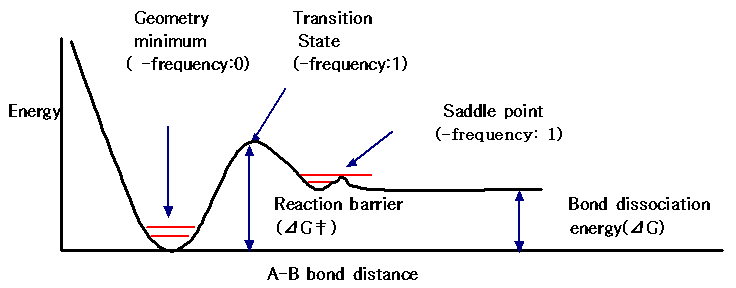
이렇게 계산되어진 전이상태를 검증하기 위해서는 verify transition state로 계산하여 IR 스펙트럼을 이용하여야 합니다. 실험적으로 IR data에는 음의 값을 갖는 frequency는 나오지 않지만, 양자화학 계산에서는 IR frequency를 계산하여 -값의 frequency가 1개 존재하면 이것을 안장점(saddle point)으로 간주합니다. 안장점(saddle point)은 한 방향을 제외하고, 다른 모든 방향으로 원자가 이동할 때 에너지가 증가하는 지점을 말합니다. 이것은 즉 분자의 힘 상수(force constant)중 오직 한 개만 음의 값을 나타내고 나머지는 모두 양의 값을 나타냄을 의미합니다. 또한 음의 힘상수를 나타내는 기준 방식(normal mode)의 진동은 음의 frequency를 나타내게 됩니다. 안장점은 2개의 극소점를 연결하는 경로 선상에서 가장 높은 에너지 상태에 있으며, 이들 극소점들은 반응물이나 생성물이 아닐 수도 있습니다. 반응 좌표(reaction coordinate)는 전이구조로부터 에너지의 감소가 가장 큰 방향으로 진행 됩니다. 아래의 그림으로 설명할 수 있는데, 화학반응에서 A-B의 결합이 길어지면서 에너지가 증가하게되고 전이상태에 도달되었을 때 가장 높은 값을 갖게 됩니다. 전이상태에서는 A-B의 결합길이에만 아래와 같은 에너지를 나타내므로 1개의 음의 frequency를 나타내게 됩니다.
만일 2개 이상의 음의 frequency가 나오면 여러 방향으로 원자들이 이동할 때 에너지가 감소할 수 있으므로 진정한 전이구조는 아닙니다. 또한 음의 frequency가 없으면 그 구조는 극소점임을 나타냅니다.
전이상태 확인은 Experiment New에서 Property of는 reaction and transition states로, Property는 verify transition state로 Using은 MOPAC PM3 FORCE로 계산을 합니다.
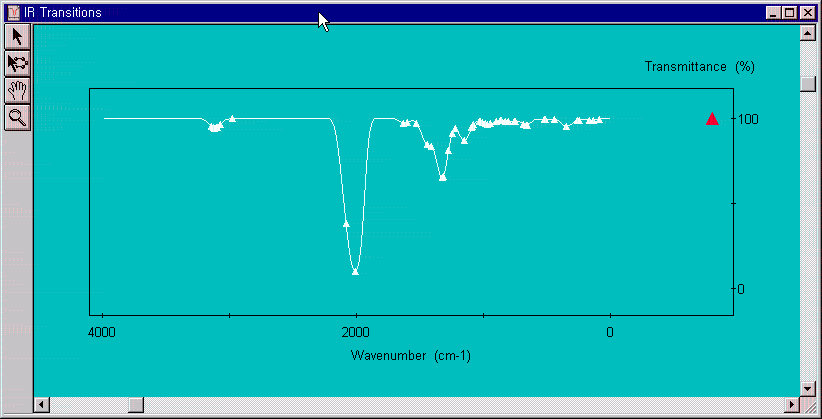
계산이 끝나고 View 메뉴안의 IR transitions을 선택하면 아래 그림과 같이 IR 스펙트럼을 볼수 있다. 이때 1개의 음의 frequency가 있음을 확인할 수 있고, 이 구조가 전이상태임을 검증할 수 있습니다. 해당하는 frequency 점을 선택하면 구조가 그려진 원도우에 해당하는 frequency의 진동방향을 볼 수 있습니다.
3.2.4. 전이상태를 알고 있는 유사한 반응을 변형하여 그 구조를 refine하는 방법
전이상태를 알고 있는 유사 반응을 이용하여 전이 상태를 찾는 방법은 앞서 계산하였던 SN2반응에서 원자를 치환하여 전이상태를 찾는 과정을 예로 설명합니다. .
Br–를 F–로 바꾸고 앞에서의 과정을 반복하여 수행합니다.
계산된 뒤에 결합 차수를 확인하고, IR frequency에서 1개의 (-) frequency가 있는지 확인합니다.
마지막으로 반응물의 생성열과 전이상태 구조에서의 생성열을 비교하여 활성화 엔탈피를 계산하도록 합니다.
3.2.5. MOPAC에서의 Saddle 계산
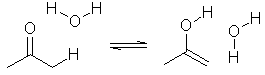
첫 부분에 설명했던 3번째의 방법으로 반응물과 생성물 두 구조를 입력하여 전이상태를 찾는 방법입니다. 예로서 아세톤의 keto-enol 토토머화(tautomerization)을 이용하도록 하겠습니다.
먼저 반응물로 acetone을 그리도록 keto라는 이름으로 저장합니다.
새로운 원도우를 열고 Copy & paste를 이용하여 acetone의 enol 형태를 저장합니다.
이때 산소에 연결하는 수소는 탄소에 연결되어있는 가장 가까운 수소와 결합으로 연결하도록 하고, 탄소와 수소와의 결합을 제거하고 C-CH 결합을 이중결합으로 바꾸도록 합니다.
이 과정은 반응물과 생성물의 atom numbering이 같도록 하기 위한 것이며 그것은 이 계산이 성공인 결과를 얻는데 있어 매우 중요합니다.
acetone의 enol구조는 Beautify Comprehensive를 실행시킨 후에 enol이라는 이름으로 저장합니다.
다음에 다시 keto구조를 t-ketoenol이라는 새이름으로 저장하도록 합니다. 이것은 saddle 계산을 수행하면 반응물의 구조가 전이상태의 구조로 바뀌어지기 때문에 원래의 반응물 구조를 보전하기 위해서입니다.
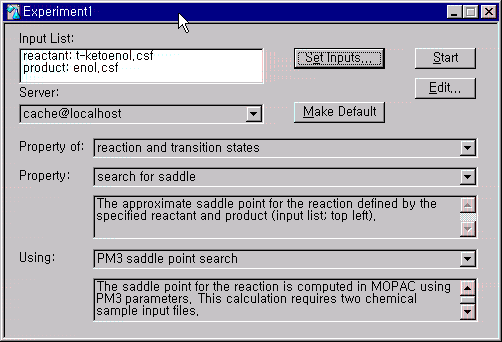
Experiment New를 선택하고 아래와 같은 원도우가 나타나는데, Property of는 reaction and transition states로, Property는 search for saddle로, Using PM3 saddle point search를 선택하도록 합니다. Input List를 보면 다른 계산방법과는 달리 reactant와 product파일을 지정해주어야 하는데, Set Inputs을 이용하여 reactant를 t-ketoenol.csf로 product는 enol.csf로 지정하고 계산을 시작합니다.
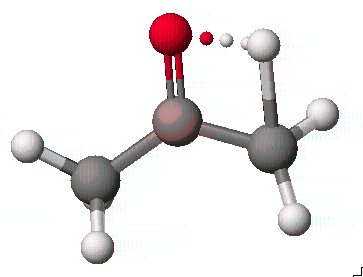
계산이 완료되면 t-ketoenol.csf구조가 그려진 원도우에는 전이 상태로 예상되는 구조로 전환되어 있습니다. 이것을 저장하고 다시 Experment New를 실행시켜 Property of는 reaction and transition states로, Property는 refine transition state로, Using은 PM3 transition state geometry로 선택하고 구조를 refine합니다.
전이상태의 생성열이 대략 21 kcal/mol인지를 확인합니다.
또한 타당한 전이상태인지를 확인하기 위하여 그 구조의 부분 결합 차수(partial bond order)를 보도록 합니다.
IR 스펙트럼을 이용한 전이상태 확인과정은 Experiment New를 선택하고 Property of는 reaction and transition states로, Property는 verify transition state로, Using은 MOPAC PM3 FORCE로 선택한 뒤 계산을 시작합니다.
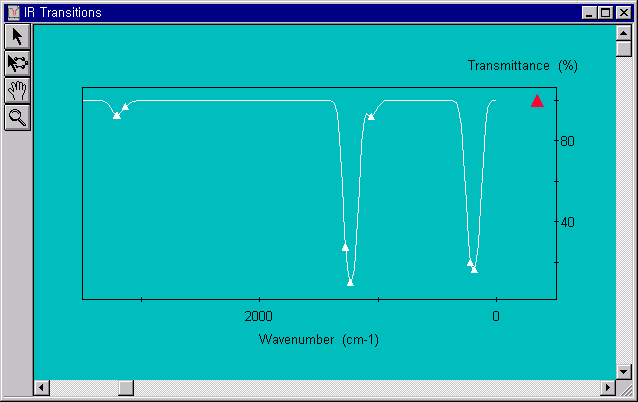
아래의 그림에서 처럼 대략 -2500cm-1에서 (-) frequency를 확인 할 수 있으므로 전이상태임을 증명할 수 있습니다.
이제 반응물(keto)과 생성물(enol)의 구조를 PM3방법으로 최적화시켜서 생성열을 계산하고 활성화 엔탈피를 결정하도록 합니다.
반응물 : keto
생성물 : enol
3.2.6. MOPAC에서의 Saddle 계산의 응용
다음 단계는 water가 촉매로 작용하는 6-membered ring 전이상태의 활성화 엔탈피를 계산하여 위의 결과와 비교해 보도록 합니다.
또한 Urethane의 고분자 반응을 이 방법으로 적용해 보도록 합니다.
먼저 methanol과 methyl isocyanate(CH3-N=C=O)을 반응물로 그리고, 이들의 생성물인 CH3-NH-(-C=O)-O-CH3를 그리고 저장합니다. 앞에서 keto-enol 토토머화(tautomerization)과정과 같은 방법으로 진행을 하며, 반응물과 생성물의 atom numbering이 변하지 않도록 유지되도록 주의합니다. 그리고 반응물의 경우 다른 이름으로 저장해 놓는 과정을 잊지 않기 바랍니다.
전이 상태의 생성열이 대략 -29kcal/mol정도 나오는지 확인하도록 합니다.
조금더 실질적인 면으로 접근해 보려면, OH의 수소에 붙어서 촉매작용을 하는 trimethylamine의 구조를 그려서 위의 결과로 얻어진 전이상태의 구조를 약간 변형하여 계산해 봅니다.
3.2.7. MOPAC에서 최적화된 격자(Optimized grid) 방법
최적화된 energy map을 이용하여 전이상태를 찾는 방법입니다.
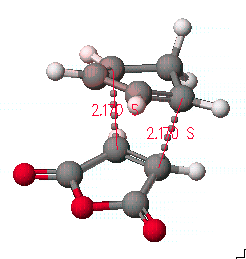
본 과정에 이용할 예는 Diels-Alder reaction중에서 cyclopentadiene 과 maleic anhydride의 반응을 알아보겠습니다.
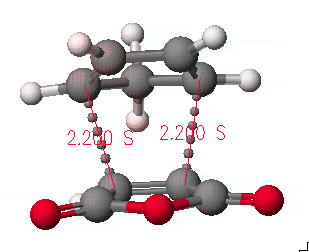
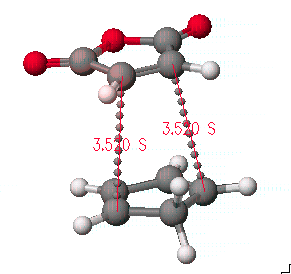
이 화합물의 합성과정은 일반적으로 diene과 dienophile사이의 π전자에 의한 상호작용으로 endo형태가 주 생성물로 알려져 있습니다. 그러므로 전이상태의 구조를 endo 형태를 중심으로 나타내도록 합니다. 그리고 Diels-Alder reaction에서는 2개의 반응중심(reaction center)에서 동시에 일어나는 반응임을 확인하도록 합니다.
초기구조를 그리기가 비교적 어렵기 때문에 쉽지 않고 많은 계산시간이 소요되는 단점이 있으나, 3차원 등고선을 통하여 분석을 쉽게 할 수 있다는 장점을 지니고 있습니다.
먼저 화학구조를 그리고 앞 페이지의 그림과 같이 두 반응물을 접근시킵니다.
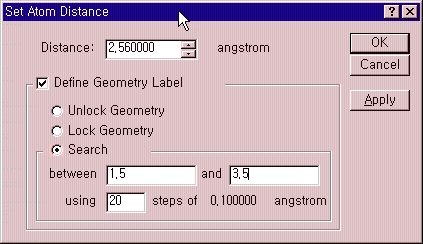
다음 과정은 반응이 일어날 원자들의 거리를 변화시킬때의 energy map을 보기 위하여, 결합이 일어날 두 원자들을 선택하고 Adjust 메뉴에서 Atom distance를 선택하면 아래의 왼쪽과 같은 원도우가 나오게 됩니다.
여기서 Define Geometry Label을 선택하고 Search에서 거리를 1.5에서 3.5사이를 0.1Å간격으로 20번 지점에서 계산하도록 설정합니다. 결합이 일어날 다른 두 원자에 대해서도 같은 설정을 해줍니다.
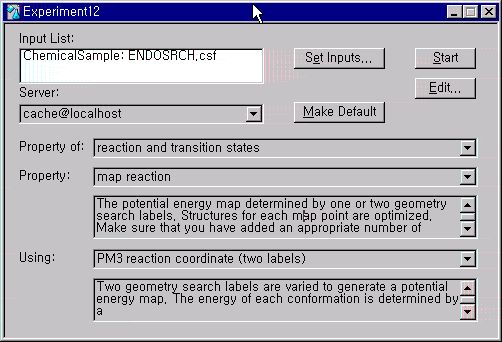
그 다음 Experiment New를 실행하고 Property of는 reaction and transition states로, Property는 map reaction으로, Using은 PM3 reaction coordinate (two lables)로 선택한 뒤 계산을 시작합니다.
아마도 이방법은 상당히 오랜시간동안 계산을 할 것입니다.
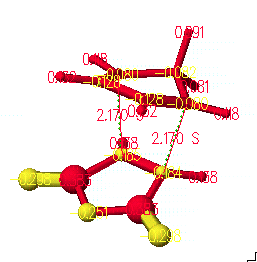
계산이 끝나면 아래와 같은 원도우가 나며 3차원 등고선위를 이동시켜 전이 상태에서의 구조를 볼 수 있습니다.
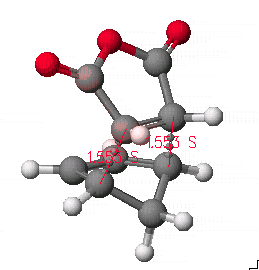
위의 결과에서 전이 상태에서의 구조를 저장하고, 조금 더 정확한 구조를 얻기 위해서는 refine transition state를 실행 시키고 계산 방법은 PM3로 합니다.
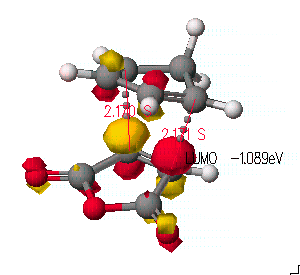
계산된 결과를 보면 전이 상태에서 원자간 거리는 2.17Å을 나타냄을 알 수 있고, 2개의 결합 길이가 같음을 알 수 있습니다.
전이 상태임을 검증하기 위하여 verify transition state를 실행하여 IR 스펙트럼을 보고 1개의 음의 frequency를 확인하도록 합니다
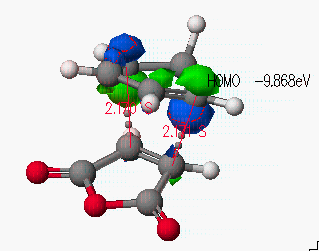
마지막으로 전이 상태의 구조에서 HOMO와 LUMO 오비탈을 보도록 하겠습니다.
아래 그림에서와 같이 endo방향으로 주 생성물이 생기는 원인을 보도록 하겠습니다.
diene의 HOMO와 dienophile의 LUMO 사이의 π전자에 의한 상호작용이 가능함을 볼 수 있으며, 이것이 endo형태가 다소 유리한 반응임을 간접적으로 설명할 수 있습니다. 조금더 정확한 증명은 exo형태의 전이구조를 결정한 후, 활성화 엔탈피를 계산함으로 증명할 수 있을 것입니다.
3.2.8. IRC를 이용한 반응좌표 결정
일단 전이 상태의 구조가 결정되면, 그 반응의 반응좌표를 결정해야 됩니다.
반응좌표를 결정하는 방법은 내성 반응좌표(Intrinsic Reaction Coordinate : 이하 IRC)방법과 동적 반응좌표(Dynamic Reaction Coordinate: DRC)방법이 있는데 여기서는 내성 반응좌표방법만 설명합니다.
IRC방법은 모든 지점에서 운동에너지(kinetic energy)를 버리고 전이구조로부터 에너지의 감소가 가장 큰 방향으로 반응경로를 계산합니다. 위치에너지(potential energy)가 변함에 따라 발생하는 운동에너지는 위치에너지와 전체 에너지를 같게 하기 위하여 억제시킵니다. IRC방법은 언제나 전이상태의 구조로부터 시작하며, 초기 이동 방향에 따라 진동방식(vibrational mode)을 결정해야 합니다. 진동방식은 mode-1과 mode+1이 있으며, 전이상태에서 반응물으로 이동하는 방향과 전이상태에서 생성물로 이동하는 가장 낮은 에너지 경로를 찾습니다. 특히 IRC방법은 반응물과 생성물 사이의 또다른 전이상태의 구조를 찾을 때 유용합니다.
먼저 3.2.7과정에서 결정된 전이상태의 구조를 이용하여 계산합니다.
전이상태의 구조에서 Experiment New를 실행시키고 Property of는 reaction and transition states로, Property는 Find reaction paths로, Using은 PM3 Intrinsic Reaction coordinate (mode=+1)로 선택한 뒤 계산을 시작합니다.
이 계산은 irc1.map이라는 파일을 만들며(만일 구조파일이름이 endo-tg.csf라면 경로는
c:\cache\user\endo-tg.map\irc1.map에 있을 것입니다.), 이것은 CAChe Workspace에서 map파일을 읽어서 그래픽으로 볼 수 있습니다.
이 과정에서 끝점이 반응물이나 생성물과 일치하는지를 확인하도록 합니다.
본 과정의 예와 다른 화합물에 대하여 적용하여 끝점이 반응물이나 생성물과 일치하지 않는다면, 그것은 전이상태의 구조를 찾은 것이 아니고 중간생성물(intermediate)의 구조를 결정한 것일 수도 있습니다.
3.2.9. Gibbs 자유 에너지
전이 상태의 구조가 정교화(refine)되면, Gibbs 자유 에너지나 엔트로피와 같은 열역학적인 데이터 등을 얻어 낼 수 있습니다. 이러한 데이터는 IR 스펙트럼을 얻어내는 과정에서 계산됩니다.